The PRALINE database
Here we introduce PRALINE - Protein and Rna humAn singLe-nucleotIde-variaNts in condEnsates - a database to interrogate proteins and RNAs contained in human condensates such as stress-granules (SGs), processing bodies (PBs), droplets, amyloids and many more.. For proteins, PRALINE reports the liquid-liquid phase separation (LLPS) and liquid-solid phase separation (LSPT) propensities as well as the predicted and experimentally validated protein and RNA interactions. For RNAs, PRALINE reports the secondary structure content, as well as the predicted and experimentally validated protein and RNA interactions. All the information is also provided considering the presence of human disease-related variants (SNVs). Where available, experimental information on the physical state (LLPS and LSPT) is given with links to relevant literature. All the predictions reported in PRALINE are based on several tools developed in our group in >15 years of activity (e.g., 18514226, 21623348, 27899588, 27320918). The algorithms are widely employed by the scientific community.
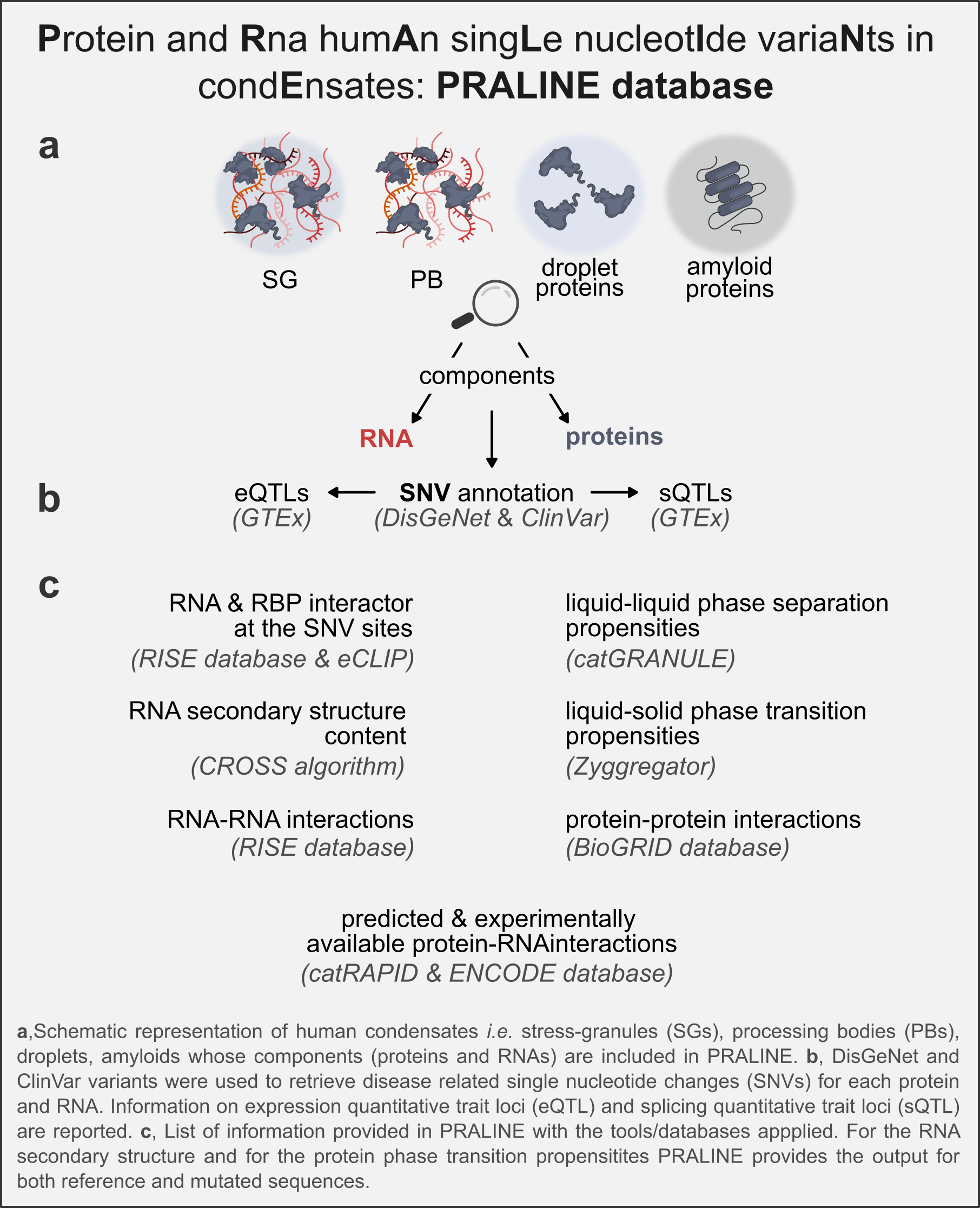
RNA molecules
All RNAs sequences were retrieved from Ensembl version 105 (34791404). 1841 Stress Granule (SG) enriched RNAs were taken from (29129640) selecting the ones with log2(fold-change) >=1 and p-value <=0.01. 4852 Processing Body (PB) enriched RNAs are taken from (28965817), retrieving the ones with log2(fold-change) >=1 and q-value <= 0.01) for a total of 5614 unique genes upon removal of obsolete gene ids in the ensembl version 105. For each gene, the longest isoform was taken. The CROSS algorithm for secondary structure prediction of RNAs was launched on the 5614 RNA sequences with Globalscore parameter.
Protein molecules
997 UniprotKB IDs were retrieved from different sources. Stress Granule (SG) proteins were retrieved from Markmiller et al. (29373831), Gingras et al. (29395067) and Jain et al. (26777405) studies. From Jain work we retrieved 411 proteins that were either already known SG proteins or newly discovered through mass spec and IF. From Markmiller and colleagues, we retrieved 397 proteins either already known from previous experiments or found with APEX technique in hek293, NPC and IPSC cells, as well as proteins found to be stress / cell specific or independent. 60 SG proteins were retrieved from Gingras work. We retrieved proteins with Non-negative matrix factorization (NMF) values = 9 or in case of a different NMF value, we collected proteins found to co-localise with G3BP1.
Processing Body (PB) proteins were retrieved from Hustemberger et al. (28965817) and Gingras et al. studies (29395067). From the hustemberger study, we collected 125 proteins found to be significantly enriched in PBs with p-value < 0.025 as reported in the paper. From Gingras and colleagues' work, we retrieved 42 proteins either with NMF value =8 or that co-localize with DCP1A.
From Vendruscolo and Fuxreiter work (34391803) we retrieved 280 droplet forming proteins and 68 amyloid forming ones.
SNVs
DisGeNet curated variant-disease associations (31680165) and ClinVar variants (24234437) with a review status higher than one (“practice guideline”, “reviewed by expert panel” and “criteria provided, multiple submitters, no conflicts”) were used to retrieve disease related single nucleotide changes (SNVs). 13857 SNVs from DisGeNet and 48671 SNVs from ClinVar fell in the coding region of RNAs enriched in SG and PB and their relative position in the transcripts was retrieved from the Ensembl Variation 105 (34791404) in Human Short Variants dataset, excluding insertions and deletions. CROSS algorithm with Globalscore parameter was launched on RNA fragments of 51 nt with the SNV in the center to calculate the difference in secondary structure content between the reference and alternative RNA sequences. To avoid smaller-size fragments, SNVs falling at the beginning and at the end of the RNAs were removed. The reference and alternative CROSS secondary structure propensity profiles of the 51 nt RNA fragments was represented against each other in plots. The mean difference in secondary structure between reference and alternative sequence was computed on a window size of 11 nt upstream and downstream the SNV. Information on the expression quantitative trait loci (eQTL) and splicing quantitative trait loci (sQTL) of the SNVs was obtained from GTEx V8 (32913098).
RNA-RNA interactions
We retrieved human RNA-RNA interactions from RISE database focusing on the experimental interactions obtained with PARIS technique. For each RNA, binding site location were mapped to the longest transcript in Ensembl 105, using blastn algorithm. We retrieved a total of 25.232 RNA-RNA interactions with at least one interactor being an enriched RNA in SG or PB. In 934 of those interactions we found at least a SNV located inside a binding site.
Protein-RNA interactions
We provide experimentally determined protein-RNA interactions validated through eCLIP experiments (32728246) as well as catRAPID predictions available in RNAct (30445601). For the experimentally validated interactions, binding sites are displayed.
Protein-Protein interactions
As binding sites related to protein-protein interactions are not physically available, we only provide links to an external database. Human curated protein-protein interactions are linked to BioGRID database (version 4.4 - 33070389).
LLPS and LSPT Propensities
We computed the propensity to undergo liquid-like and solid-like condensation for the set of 997 proteins detailed previously and for their 6152 natural variants (involving 632 of them), retrieved from UniProtKB. We considered 5949 single point mutations and 203 deletions. To quantify the extent to which each sequence is prone to undergo LSPT and LLPS we used the Zyggregator and catGRANULE algorithms, which compute the liquid-solid and liquid-liquid propensities, respectively.
LLPS and LSPT are promoted by both intrinsic and extrinsic contributions. By intrinsic contributions we mean physico-chemical properties of the polypeptide chain such as the hydrophobicity for Zyggregator and the structural disorder for catGRANULE. The extrinsic contributions relate to environmental factors, such as concentration, pH, ionic strength, but not only, for instance crowding agents are also to be included. Our methods only predict intrinsic properties of the polypeptide chain and do not take into account the different extrinsic contributions at present.
In the figure we show our LLPS and LSPT predictions considering a reference data set composed of amyloid prone regions APR (29040693) and droplet prone regions DPR (35025997 and 34391803)
For the proteins that do not have natural variants present in UniProtKB we only report the WT scores of Zyggregator and catGRANULE.
We note that while Zyggregator has been intensively tested on different protein mutants, there is not a database to benchmark catGRANULE performances on individual SNVs.
Currently, there is a lack of experimental data to precisely determine the ability of our method. Yet, not only have we thoroughly tested our algorithm on the human and yeast proteome (30566867),
but we also successfully based our experimental mutational assays on its predictions (27320918 and 32857852).
Indeed, in Figure 5 of the manuscript in which we introduced catGRANULE (27320918),
we designed a set of mutants whose phase separation propensity was assessed in cells (see also Supplementary Figure 5) and showed excellent agreement with the predictions.
Similarly, in Figure 3 of a follow-up work (32857852), we performed deletion of phase separating regions following our predictions and reported a great match with our calculations.
This approach has been followed by others in high-level journals, including Nature’s paper "Assembly of synaptic active zones requires phase separation of scaffold molecules"
(Figure 1 and extended data Figure 1) (33208945) and PNAS-’s paper "Phase separation by ssDNA binding protein controlled via protein−protein and protein−DNA interactions" (Figure 1) (33020264),
which again reported great predictive power of our tool by identifying the prion domain responsible for phase separation. Thus, although few data are available, we believe that our method, even if affected by error as any algorithm, has a great potential to unveil variants involved in phase separation.
We would like to mention that the manuscript "First-generation predictors of biological protein phase separation"
shows that catGRANULE has the highest AUC in identifying known phase separating proteins and one of the highest true positive rates (31252218)"
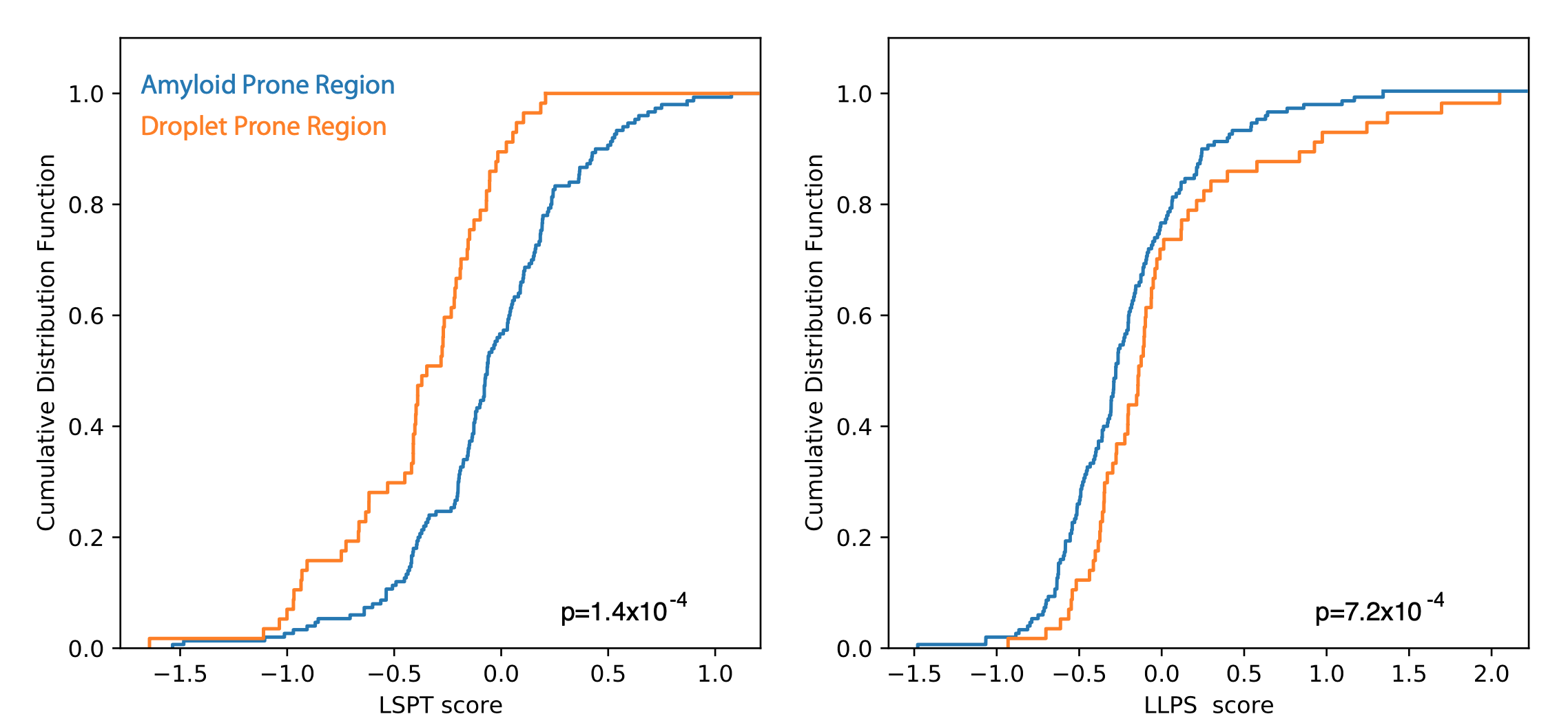 Figure 1 Cumulative Distribution Functions of LSPT and LLSP propensitives for experimentally-derived Amyloid-prone (blue) and Droplet-prine (oraange) protein regions. Statistics show that Amyloid and Droplet prone regions can be correctly distinguished with a p-value of 1.4 x 10^-4 and 7.2 x 10^-, respectively.
We note that droplets can evolve towards the amyloid state, while the opposite does not occur.
Figure 1 Cumulative Distribution Functions of LSPT and LLSP propensitives for experimentally-derived Amyloid-prone (blue) and Droplet-prine (oraange) protein regions. Statistics show that Amyloid and Droplet prone regions can be correctly distinguished with a p-value of 1.4 x 10^-4 and 7.2 x 10^-, respectively.
We note that droplets can evolve towards the amyloid state, while the opposite does not occur.
Funding
The research leading to these results has been supported by European Research Council [RIBOMYLOME_309545 and ASTRA_855923] and the H2020 projects [IASIS_727658 and INFORE_825080].
Acknowledgements
- The authors thank Adriano Setti for the discussion about the RNA-RNA interactions section and Leila Mansouri that came up with the database’s name.
- Template and CSS from Bootstrap.
Licence
Our own work is licenced under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International Licence .
.
© 2022 tartaglialab.com